¿Qué es el canal de cloruro regulador de conductancia transmembrana de fibrosis quística (CFTR)? – Análisis Completo y Beneficios
Resumen Clínico Rápido
🔬 Clasificación
Canal Iónico, Transportador ABC
⚙️ Función
Transporte de Cloruro y Bicarbonato, Regulación de Fluidos
📋 Impacto
Causa principal de Fibrosis Quística
¿Qué es el Canal de Cloruro Regulador de Conductancia Transmembrana de Fibrosis Quística (CFTR)?
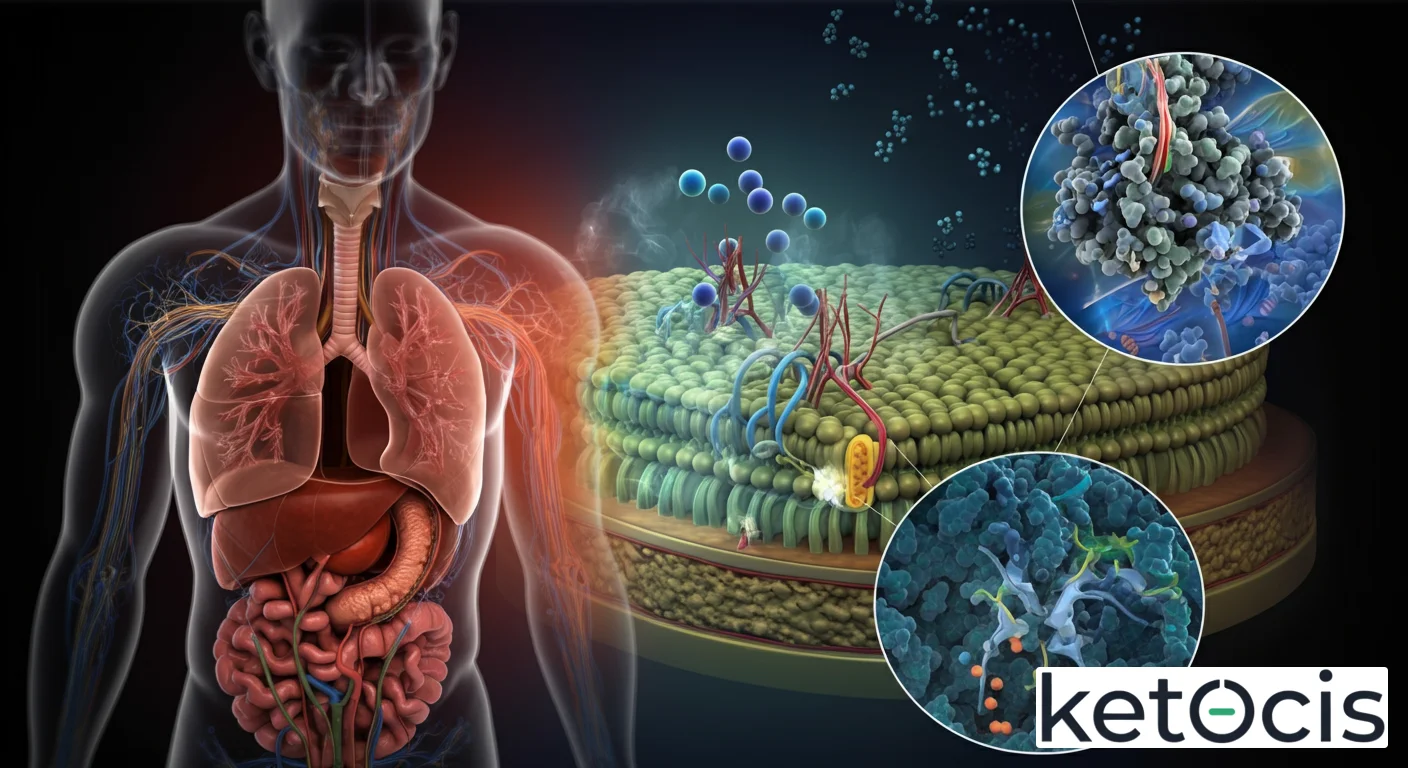
El nombre completo, Canal de Cloruro Regulador de Conductancia Transmembrana de Fibrosis Quística, puede parecer intimidante, pero se refiere a una proteína de importancia capital para la fisiología humana y, cuando disfunciona, para una de las enfermedades genéticas más devastadoras: la fibrosis quística. Conocida universalmente por sus siglas en inglés, CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), esta proteína es mucho más que un simple canal; es un sofisticado regulador del movimiento de iones y agua a través de las membranas celulares, impactando directamente la hidratación de las secreciones en múltiples órganos.
En su esencia, el CFTR es una proteína que actúa como un canal iónico, permitiendo el paso de iones de cloruro (Cl-) y, en menor medida, de bicarbonato (HCO3-) a través de la membrana apical de las células epiteliales. Esta función es crucial para mantener el equilibrio de líquidos y electrolitos, lo que a su vez asegura que las secreciones corporales, como el moco, el sudor, los jugos pancreáticos y la bilis, tengan la consistencia adecuada para fluir libremente y cumplir sus funciones biológicas. Sin un CFTR funcional, estas secreciones se vuelven anormalmente espesas y pegajosas, obstruyendo conductos y órganos, lo que es el sello distintivo de la fibrosis quística.
La comprensión del CFTR y su intrincada biología molecular ha transformado la forma en que entendemos y tratamos la fibrosis quística. Desde su descubrimiento genético en 1989, la investigación ha desvelado no solo su estructura y mecanismo de acción, sino también la vasta gama de mutaciones que pueden afectar su función, llevando a un espectro de manifestaciones clínicas. Este conocimiento ha allanado el camino para el desarrollo de terapias revolucionarias que, por primera vez, abordan la causa subyacente de la enfermedad en lugar de solo sus síntomas, ofreciendo una esperanza sin precedentes a los pacientes.
Resumen Clínico
- El CFTR es una proteína canal iónico esencial para el transporte de cloruro y bicarbonato.
- Regula la hidratación de secreciones en pulmones, páncreas, intestino, glándulas sudorosas y otros órganos.
- Las mutaciones en el gen CFTR causan la fibrosis quística, caracterizada por secreciones espesas y daño orgánico progresivo.
- Las terapias modernas con moduladores del CFTR están revolucionando el tratamiento al restaurar la función proteica.
El Descubrimiento Genético y la Estructura Molecular del CFTR
La historia del CFTR es una de las grandes epopeyas de la genética moderna. En 1989, un equipo de investigadores identificó y clonó el gen responsable de la fibrosis quística, un hito que no solo reveló la naturaleza molecular de la enfermedad, sino que también sentó las bases para el estudio intensivo de esta proteína. El gen CFTR se localiza en el brazo largo del cromosoma 7 y codifica una proteína de 1480 aminoácidos, que pertenece a la superfamilia de transportadores ABC (ATP-binding cassette).
La estructura del CFTR es compleja y altamente conservada a lo largo de la evolución, lo que subraya su importancia funcional. Se compone de cinco dominios principales: dos dominios transmembrana (TMD1 y TMD2), cada uno con seis hélices alfa que se insertan en la membrana celular y forman el poro del canal; dos dominios de unión a nucleótidos (NBD1 y NBD2), que se encuentran en el citoplasma y unen y hidrolizan ATP; y un dominio regulador (R), también citoplasmático, que es crucial para la activación del canal. Esta arquitectura modular permite una regulación precisa de su actividad, un aspecto fundamental para su función fisiológica.
Mecanismo de Acción: Un Canal Regulado por Energía y Fosforilación
El CFTR no es un canal iónico simple que se abre y cierra pasivamente. Su actividad está finamente controlada por un mecanismo que involucra tanto la unión e hidrólisis de ATP como la fosforilación. Para que el canal se active y permita el flujo de iones de cloruro, el dominio R debe ser fosforilado por la proteína quinasa A (PKA), una enzima que responde a señales intracelulares como el AMP cíclico (cAMP). Una vez fosforilado, el dominio R sufre un cambio conformacional que permite que los dominios NBD se unan al ATP.
La unión y la posterior hidrólisis de ATP en los NBDs provocan una serie de cambios conformacionales que abren y cierran el poro del canal. Este ciclo de unión-hidrólisis de ATP es lo que impulsa la apertura del canal, permitiendo que los iones de cloruro fluyan a favor de su gradiente electroquímico. Este proceso no solo mueve cloruro, sino que también influye en el movimiento de agua y otros iones, como el sodio, a través de la membrana, lo que es esencial para la hidratación de las superficies epiteliales. Por ejemplo, en las vías respiratorias, el CFTR transporta cloruro hacia el exterior de la célula, lo que atrae agua y mantiene el moco delgado y fácil de limpiar. En las glándulas sudoríparas, sin embargo, el CFTR reabsorbe cloruro de nuevo hacia la célula, lo que evita una pérdida excesiva de sal.
La fibrosis quística es solo una enfermedad pulmonar.
La fibrosis quística es una enfermedad multisistémica que afecta pulmones, páncreas, hígado, intestino, glándulas sudoríparas y el sistema reproductivo debido a la disfunción del CFTR.
CFTR y la Fibrosis Quística: Una Disfunción Multiorgánica
La fibrosis quística (FQ) es la enfermedad genética letal más común en poblaciones caucásicas, causada por mutaciones en el gen CFTR. Se han identificado más de 2000 mutaciones diferentes, pero la más prevalente es la deleción de tres pares de bases que resulta en la pérdida del aminoácido fenilalanina en la posición 508 (ΔF508). Esta mutación, y muchas otras, impiden que la proteína CFTR se pliegue correctamente, se procese adecuadamente en el retículo endoplasmático y, en última instancia, alcance la membrana celular para ejercer su función.
La disfunción del CFTR conduce a una amplia gama de manifestaciones clínicas que afectan principalmente a los sistemas respiratorio, digestivo y reproductivo:
- Sistema Respiratorio: Es la principal causa de morbilidad y mortalidad en pacientes con FQ. La falta de CFTR funcional en las células epiteliales de las vías respiratorias resulta en un moco espeso y pegajoso que atrapa bacterias (como Pseudomonas aeruginosa y Staphylococcus aureus) y es difícil de eliminar. Esto conduce a infecciones crónicas, inflamación persistente, daño pulmonar progresivo (bronquiectasias) e insuficiencia respiratoria.
- Sistema Digestivo: La obstrucción de los conductos pancreáticos por secreciones espesas impide que las enzimas digestivas lleguen al intestino. Esto causa insuficiencia pancreática exocrina, lo que lleva a una malabsorción severa de grasas y vitaminas liposolubles (A, D, E, K), desnutrición y retraso del crecimiento. Muchos pacientes desarrollan también diabetes relacionada con FQ (DRFQ) debido al daño a las células productoras de insulina en el páncreas. En el intestino, el moco espeso puede causar obstrucciones, como el íleo meconial en recién nacidos o el síndrome de obstrucción intestinal distal (SOID) en adultos.
- Glándulas Sudoríparas: A diferencia de otros órganos, en las glándulas sudoríparas, el CFTR reabsorbe cloruro del sudor primario antes de que llegue a la superficie de la piel. En la FQ, esta reabsorción es defectuosa, lo que resulta en un sudor con un contenido anormalmente alto de sal. Esta característica es la base del «test del sudor», una prueba diagnóstica clave para la FQ.
- Sistema Reproductivo: La mayoría de los hombres con FQ son infértiles debido a la ausencia o bloqueo del conducto deferente (azoospermia obstructiva), que transporta los espermatozoides. Las mujeres con FQ pueden experimentar una fertilidad reducida debido a las secreciones cervicales espesas.
- Hígado: La bilis espesa puede bloquear los conductos biliares, llevando a cirrosis biliar y enfermedad hepática.
Diagnóstico y Manejo de la Fibrosis Quística
El diagnóstico temprano de la FQ es crucial para iniciar el tratamiento y mejorar los resultados a largo plazo. En muchos países, el cribado neonatal (prueba del talón) detecta niveles elevados de tripsinógeno inmunorreactivo (IRT), lo que sugiere la necesidad de pruebas adicionales. La prueba definitiva suele ser el test del sudor, que mide la concentración de cloruro en el sudor. Un nivel elevado de cloruro es altamente indicativo de FQ. La confirmación genética mediante el análisis de mutaciones del gen CFTR es también una herramienta diagnóstica esencial.
Terapias Revolucionarias: Moduladores del CFTR
Durante décadas, el tratamiento de la FQ se centró en el manejo sintomático: fisioterapia respiratoria para limpiar las vías aéreas, antibióticos para las infecciones, enzimas pancreáticas para la malabsorción y suplementos nutricionales. Si bien estas terapias mejoraron la calidad de vida y la esperanza de vida, no abordaban la causa subyacente de la enfermedad. La verdadera revolución llegó con el desarrollo de los moduladores del CFTR.
Estos fármacos actúan directamente sobre la proteína CFTR defectuosa para restaurar su función:
- Potenciadores (ej. Ivacaftor): Mejoran la función de las proteínas CFTR ya presentes en la superficie celular, ayudándolas a abrirse más frecuentemente y permitiendo un mayor flujo de cloruro. Son efectivos para mutaciones que resultan en una proteína CFTR presente pero disfuncional (clase III y IV).
- Correctores (ej. Lumacaftor, Tezacaftor, Elexacaftor): Ayudan a que las proteínas CFTR mutadas se plieguen correctamente y se transporten a la superficie celular. Son cruciales para la mutación ΔF508 (clase II), donde la proteína se degrada antes de llegar a su destino.
- Amplificadores y Agentes de Lectura (Read-through agents): Actualmente en investigación, buscan aumentar la cantidad de proteína CFTR producida o permitir que los ribosomas «lean» a través de mutaciones sin sentido (clase I) para producir una proteína funcional.
Las terapias combinadas, como los tratamientos de triple combinación (ej. Elexacaftor/Tezacaftor/Ivacaftor, conocido como Trikafta o Kaftrio), han transformado el panorama de la FQ para la mayoría de los pacientes con la mutación ΔF508. Estos tratamientos han demostrado mejoras dramáticas en la función pulmonar, el estado nutricional y la calidad de vida, extendiendo significativamente la esperanza de vida y aliviando la carga de la enfermedad.
Biohacking y Optimización Celular para la Función del CFTR
Aunque los moduladores del CFTR son el pilar del tratamiento, la optimización celular a través de hábitos de vida puede complementar la salud general. Una hidratación adecuada es fundamental para mantener la fluidez de las secreciones. Además, dietas ricas en antioxidantes y antiinflamatorios, junto con ejercicio regular, pueden mitigar el estrés oxidativo y la inflamación crónica asociados a la FQ, apoyando la salud pulmonar y general, aunque no reemplazan la terapia farmacológica específica para el CFTR.
Más Allá de la Fibrosis Quística: Otras Conexiones del CFTR
Si bien la FQ es la enfermedad más conocida asociada con el CFTR, la disfunción de esta proteína puede contribuir a otras afecciones, a menudo de forma más sutil o en casos de mutaciones más leves. Estas incluyen:
- Pancreatitis Idiopática Recurrente: Algunas mutaciones del CFTR, incluso en ausencia de FQ completa, pueden predisponer a episodios recurrentes de inflamación pancreática.
- Bronquiectasias no relacionadas con FQ: Pacientes con bronquiectasias crónicas pero sin un diagnóstico de FQ pueden tener mutaciones leves o variantes del CFTR que afectan parcialmente la función pulmonar.
- Azoospermia Obstructiva Congénita Bilateral del Conducto Deferente (CBAVD): Es una causa común de infertilidad masculina, y la mayoría de los hombres con CBAVD tienen al menos una mutación del CFTR, incluso si no presentan otros síntomas de FQ.
Estos ejemplos resaltan la versatilidad y la importancia del CFTR en la fisiología de múltiples sistemas orgánicos, y cómo incluso una disfunción parcial puede tener implicaciones clínicas significativas.
Alerta Médica: Mitos sobre la Curación de la Fibrosis Quística
Es un mito peligroso creer que cambios dietéticos extremos, suplementos «milagrosos» o terapias no probadas pueden «curar» la fibrosis quística o reemplazar los tratamientos médicos actuales. La FQ es una enfermedad genética compleja que requiere un manejo médico integral y, cuando sea aplicable, terapias moduladoras del CFTR aprobadas. Abandonar el tratamiento médico basado en afirmaciones pseudocientíficas puede tener consecuencias devastadoras para la salud del paciente.
El Futuro del Tratamiento y la Investigación del CFTR
El campo de la investigación del CFTR es uno de los más dinámicos de la medicina moderna. Los avances recientes en terapias moduladoras son solo el comienzo. Las líneas de investigación futuras incluyen:
- Terapia Génica y Edición Genómica: El objetivo es corregir el gen CFTR defectuoso directamente en las células del paciente utilizando técnicas como CRISPR/Cas9 o la entrega de genes funcionales a través de vectores virales. Esto podría ofrecer una cura definitiva, especialmente para aquellos pacientes que no responden a los moduladores actuales.
- Terapias de ARNm: El desarrollo de terapias basadas en ARN mensajero (ARNm) para producir la proteína CFTR funcional, similar a las vacunas de ARNm, es una vía prometedora.
- Medicina Personalizada: Continuar desarrollando terapias dirigidas a mutaciones específicas, explorando combinaciones de moduladores y fármacos que aborden las características únicas de cada paciente.
- Modelos de Enfermedad Avanzados: El uso de organoides (cultivos de células 3D que imitan la estructura y función de órganos) derivados de pacientes con FQ está permitiendo probar nuevos fármacos y entender mejor la patofisiología de la enfermedad.
El CFTR ha pasado de ser un misterioso gen a un objetivo terapéutico que ha reescrito la historia natural de una enfermedad devastadora. Su estudio no solo ha beneficiado a los pacientes con fibrosis quística, sino que también ha proporcionado conocimientos invaluables sobre la fisiología de los canales iónicos, el plegamiento de proteínas y el desarrollo de fármacos de precisión. La continua investigación promete seguir desvelando los secretos de esta fascinante proteína y ofrecer aún más esperanza para el futuro.
Preguntas Frecuentes Relacionadas
¿Cuánto tiempo tarda el proceso metabólico?
El tiempo varía según el metabolismo individual y la adherencia a la restricción de carbohidratos, pero generalmente toma de 2 a 4 días en condiciones estrictas.
¿Cómo mido mis niveles de forma óptima?
Se recomiendan los medidores de sangre para mayor precisión clínica (miden beta-hidroxibutirato), aunque existen opciones de aliento y tiras de orina para principiantes.
¿Es normal sentir fatiga al inicio?
Sí, durante la fase de adaptación es común experimentar la «gripe keto». Mantener una óptima hidratación y reponer electrolitos (sodio, potasio, magnesio) mitiga drásticamente estos efectos.
Explorar Glosario Médico
Explora Nuestros Centros Temáticos
Tu Panel Metabólico
Sincroniza tus registros y monitorea tu progreso en tiempo real con Ketocis Tracker.
Directorio de Recetas Keto
Explora nuestra base de datos completa de platillos bajos en carbohidratos.